Một vài nét về bệnh AMDM
Acromesomelic Dysplasia (AMD) là một bệnh di truyền hiếm gặp (theo Hiệp hội bệnh hiếm châu Âu, bệnh hiếm là bệnh với tỷ lệ gặp thấp hơn 1/2000 người) gây ra bởi đột biến gen lặn trên nhiễm sắc thể thường, dẫn đến kiểu hình thấp lùn và bất thường phát triển xương tại các vị trí cẳng tay, cẳng chân và bàn tay, bàn chân. Bệnh AMD gồm ba thể [Maroteaux (AMDM), Grebe (AMDG), Hunter Thompson (AMDH)], việc chẩn đoán xác định dựa vào sự khác biệt trong biểu hiện lâm sàng, X quang và quan trọng nhất là xét nghiệm gen phát hiện đột biến.
Năm 1971, Maroteaux và cộng sự phát hiện ca lâm sàng AMDM đầu tiên với các biểu hiện bất thường xương đặc trưng như: các đoạn giữa và xa của tay và chân (bao gồm cẳng tay, cẳng chân, bàn tay và bàn chân) có kích thước không đồng nhất với cơ thể. Hiện tượng này thường được nhận thấy ngay từ khi sinh và trở nên rõ ràng hơn trong 2 năm đầu đời. Ngoài ra ở bệnh nhân AMDM, cân nặng và chiều cao lúc sinh bình thường, nhưng một số hiện tượng xương dài ngắn hơn bình thường có thể phát hiện ngay qua Xquang. Năm 1998, Kant và cộng sự nghiên cứu 18 gia đình có bệnh nhân AMDM và lần đầu tiên xác định được vị trí gen NPR2 là 9p13-q12 trên nhiễm sắc thể số 9. AMDM là bệnh lý di truyền đơn gen, do đó giải trình tự gen NPR2 có vai trò quan trọng trong chẩn đoán xác định bệnh. Tuy nhiên, đột biến gen NPR2 có thể làm giảm hoặc tăng chức năng của gen dẫn đến kiểu hình lùn hoặc cao bất thường.
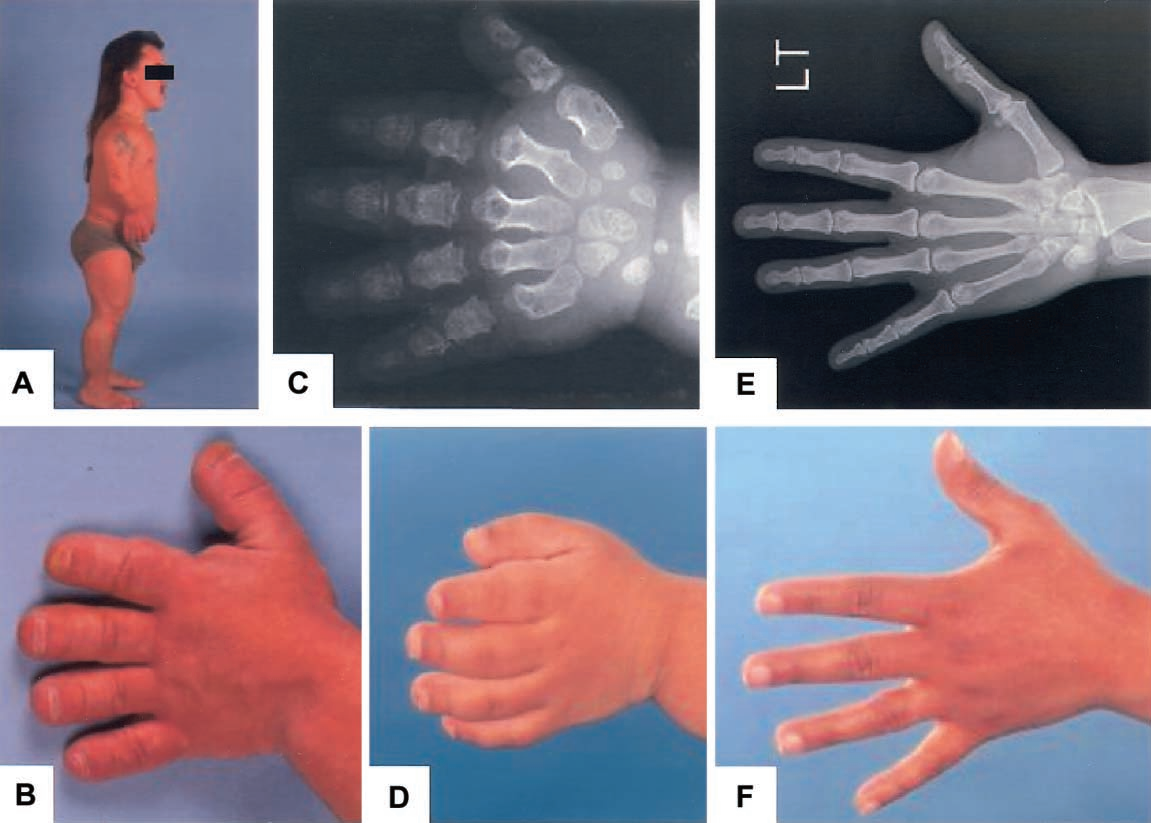
Hình 1. Hình ảnh lâm sàng và Xquang bệnh nhân AMDM.
Quá trình điều trị bệnh AMDM cần sự đóng góp của một nhóm các chuyên gia thuộc nhiều lĩnh vực khác nhau như: nhi, nội cơ xương khớp, phục hồi chức năng, di truyền học. Đối với các bất thường cột sống, dẫn đến bệnh lý như hẹp lồng ngực, biến dạng đốt sống thắt lưng có thể được điều trị bằng sự kết hợp giữa bài tập chức năng và thể thao, cùng với các kỹ thuật hỗ trợ, nặng nhất có thể phải phẫu thuật. Một số can thiệp sớm khác cũng nên được thực hiện nhằm giúp các trẻ mắc AMDM có thể tìm ra năng lực và phát triển toàn diện bản thân. Ngoài ra, tư vấn di truyền cũng là một vấn đề cần chú trọng, đặc biệt với gia đình đã sinh con được chẩn đoán AMDM, và các thành viên họ hàng có mang gen gây bệnh.
Hiện nay bệnh lý di truyền hiếm ở Việt Nam và trên thế giới đang được đi sâu quan tâm nghiên cứu. Mạng lưới bệnh hiếm và ngày bệnh hiếm trên thế giới đã được thành lập với mục đích giúp cho các bác sỹ cũng như người dân nâng cao nhận thức về tầm quan trọng của bệnh hiếm. Nghiên cứu “Phát hiện đột biến trên gen NPR2 và ứng dụng trong chẩn đoán tiền làm tổ bệnh AMDM” của chúng tôi nhằm góp phần vào tình hình nghiên cứu chung của bệnh hiếm tại Việt Nam cũng như làm tiền đề cho những nghiên cứu tiếp theo về lĩnh vực bệnh này ở nước ta.
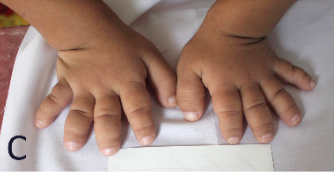
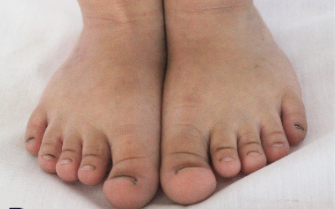
Hình ảnh bàn chân, bàn tay bệnh nhân tham gia nghiên cứu.
Phát hiện đột biến mới gây bệnh AMDM
Nghiên cứu được thực hiện trên 2 bệnh nhân (8,4 và 4,9 tuổi) là chị em gái đã được chẩn đoán xác định bệnh AMDM và thành viên gia đình bệnh nhân (bố, mẹ, ông, bà…). Phương pháp nghiên cứu được thực hiện như sau: 2 bệnh nhân được thăm khám và thực hiện xét nghiệm thường quy để loại trừ bệnh lý gây lùn hay gặp như lùn tuyến yên; thu thập mẫu bệnh phẩm của 2 bệnh nhân, thực hiện khuếch đại gen NPR2 bằng PCR và giải trình tự gen bằng Sanger Sequencing để phát hiện đột biến chỉ điểm trên gen NPR2; thu mẫu bệnh phẩm của gia đình bệnh nhân, thực hiện giải trình tự gen NPR2 và phát hiện đột biến gen; thực hiện chẩn đoán tiền làm tổ bằng cách sử dụng kỹ thuật PGS để phát hiện đột biến nhiễm sắc thể và giải trình tự gen NPR2 phát hiện đột biến chỉ điểm, từ đó chọn lựa phôi phù hợp.
Kết quả khám lâm sàng chị gái phát hiện chiều dài tay và chân đều ngắn bất thường, trong đó các ngón tay và ngón chân ngắn, rộng, vuông. Hai ngón chân cái to hơn các ngón chân còn lại. Khuỷu tay có biến dạng vẹo ra ngoài. Khám đầu mặt cổ, tim mạch, hô hấp, tiêu hóa, tiết niệu, thần kinh của trẻ hoàn toàn bình thường. Chưa ghi nhận hạn chế vận động của trẻ. Với kết quả Xquang chị gái cho thấy, chiều dài của xương quay và xương trụ ngang nhau và không có di lệch đầu xương quay, tuy nhiên xương quay ở cả hai tay đều cong. Các xương đốt bàn và đốt ngón tay đều ngắn, rộng, với đầu xương hình nón, đặc biệt là đốt ngón xa. Các đốt bàn và ngón chân cũng có đặc điểm tương tự, đặc biệt là xương ngón cái to hơn hẳn các ngón chân còn lại. Không phát hiện hiện tượng liền xương hoặc khuyết xương, xương sọ, cột sống, chậu hông chưa phát hiện bất thường.
Kết quả khám lâm sàng và Xquang của em gái cũng phát hiện nhiều đặc điểm tương tự như chị gái. Bố và mẹ của hai trẻ đều cao 158 cm (-1SD với nam, +1SD với nữ).
Kết quả nghiên cứu đột biến gen NPR2 bằng kỹ thuật PCR và giải trình tự gen bằng Sanger Sequencing cho thấy, hai bệnh nhân mang đột biến đồng hợp tử T>C tại vị trí nucleotide thứ 152 (c.152 T>C) thuộc exon 1 của gen NPR2. Đây là đột biến sai nghĩa, dẫn đến sự thay thế amino acid thứ 51 từ Leucine thành Proline (p.L51P). Bố, mẹ, bác (bên nội) và bà nội mang đột biến dị hợp tử về đột biến trên.
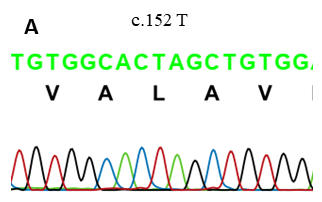
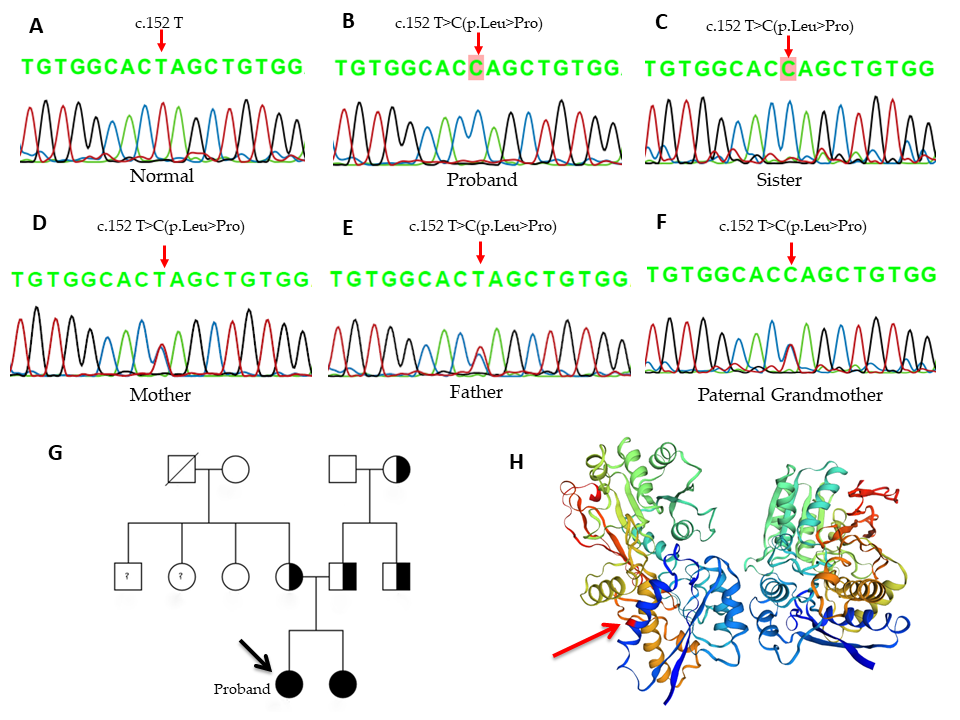
Hình 2. Kết quả giải trình tự gen NPR2. (A) của người bình thường, (B) của chị gái, (C) của em gái.
Sau khi đối chiếu với các ngân hàng đột biến gen quốc tế như Human Gene Mutation Database (http://www.hgmd.cf.ac.uk/), LOVD - Human Variome Project (https://www.lovd.nl/), ClinVar - NCBI (https://www.ncbi.nlm.nih.gov/clinvar/), chúng tôi nhận thấy, c.152 T>C là một đột biến mới, chưa từng được công bố. Các công cụ phân tích đột biến in silico đều xác nhận mức độ gây bệnh cao của đột biến c.152 T>C: Mutation Taster cho kết quả “disease causing” (gây bệnh) (http://www.mutationtaster.org/); Polyphen 2 đánh giá thang điểm gây bệnh 0,975/1 (http://genetics.bwh.harvard.edu/pph2); kết quả đánh giá của công cụ CADD là 23,8/40 (https://cadd.gs.washington.edu/), DANN là 0,9984/1 (Varsome the Human Genomics Community).
Ứng dụng thành công phát hiện đột biến mới trong chẩn đoán tiền làm tổ
Chẩn đoán tiền làm tổ (PGD) là phương pháp cho phép các cặp vợ chồng có nguy cơ sinh con mang bệnh lý di truyền có thể sinh con không mắc bệnh, dựa vào việc phát hiện các bất thường di truyền ở phôi (in-vitro) và chọn lọc phôi không có bất thường di truyền trước sinh chuyển phôi. Nhờ đó, PGD không cần phụ thuộc vào chẩn đoán trước sinh khi đã mang thai và giảm khả năng bỏ thai nếu phát hiện thai bệnh lý.
Sử dụng kỹ thuật giải trình tự gen NPR2 đã giúp xác định được bố và mẹ bệnh nhân có kiểu gen dị hợp tử về đột biến c.152 T>C. Vì AMDM là bệnh lý di truyền lặn trên nhiễm sắc thể thường nên đối với cặp bố mẹ dị hợp tử, mỗi lần mang thai, khả năng có con mắc bệnh là 25%. Một số lựa chọn sinh sản đối với cặp bố mẹ bao gồm: mang thai tự nhiên với xác suất con không bệnh là 75%; thực hiện thăm dò trước sinh khi đang mang thai để phát hiện thai nhi bất thường (chọc dịch ối, sinh thiết gai rau); hoặc chẩn đoán tiền làm tổ. Cả hai phương án đầu tiên đều gây khó khăn cho bà mẹ, do người mẹ đều phải mang thai rồi mới kiếm tra khả năng con mắc bệnh hay không. Nếu con mắc bệnh, việc đình chỉ thai nghén (bằng nạo, hút thai) không chỉ ảnh hưởng đến tinh thần và sức khỏe sản phụ mà còn có thể làm giảm khả năng thụ thai tiếp theo. Trong khi đó, chẩn đoán tiền làm tổ đảm bảo tỷ lệ cao bà mẹ không mang thai bệnh lý do kết hợp giữa thụ tinh nhân tạo và các kỹ thuật xét nghiệm di truyền sàng lọc phôi bệnh trước khi bà mẹ mang thai. Ứng dụng kết quả nghiên cứu vào chẩn đoán tiền làm tổ đã xác định được 4 phôi đạt tiêu chuẩn không mang đột biến nhiễm sắc thể và đột biến gen NPR2. Sau khi chọn lựa được phôi và được sự tư vấn di truyền của các bác sỹ, gia đình bệnh nhân đã quyết định sinh tiếp. Tin mừng đã đến với gia đình bệnh nhân vào cuối năm 2019, khi người mẹ sinh bé trai khỏe mạnh và chưa phát hiện dấu hiệu bệnh lý AMDM.