SARS-CoV-2 Mpro
Các virus corona có bộ gen lớn nhất trong số tất cả các virus RNA đã biết, chúng có chiều dài từ 26-32 kb, mã hóa các protein cấu trúc và phi cấu trúc. Bộ gen SARS-CoV-2 mã hóa hơn 20 protein, bao gồm cả enzyme protease chính (main protease - Mpro), có tỷ lệ tương đồng rất cao với enzyme protease tương ứng của virus SARS-CoV (gây hội chứng hô hấp cấp SARS) [1]. Mpro đóng vai trò quan trọng trong phiên mã và nhân bản virus SARS. Khi RNA thông tin của virus được phiên mã sẽ tạo ra các phân tử protein kết hợp (polyprotein), Mpro sẽ phân tách polyprotein thành các protein thành phần, cần thiết cho quá trình nhân bản của virus [2]. Nếu Mpro không hoạt động, quá trình nhân bản của virus sẽ bị ảnh hưởng nặng nề. Do đó, SARS-CoV-2 Mpro được xác định là mục tiêu hàng đầu để nghiên cứu thiết kế thuốc. Đến nay, một số chất ức chế đã được thiết kế để tấn công enzyme này (hình 1). Mặc dù đã có nhiều loại thuốc được phê duyệt khẩn cấp để điều trị COVID-19 [3], nhưng nhiều loại không thực sự đem lại hiệu quả, thậm chí khi thử nghiệm lâm sàng còn có kết quả thất vọng [4]. Mặc dù một số vaccine đã được đưa vào sử dụng và phát huy hiệu quả nhưng vẫn còn nhiều khó khăn trong việc cung cấp vaccine trên toàn thế giới. Do vậy, việc tìm kiếm một phương pháp điều trị thích hợp đối với đại dịch COVID-19 là rất cấp thiết.
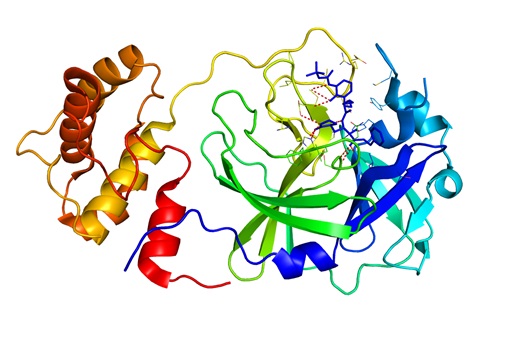
Hình 1. Cấu trúc nhiễu xạ tia X của chất ức chế α-ketoamide 13b liên kết với SARS-CoV-2 Mpro (PDB ID:6Y2F).
Sử dụng máy tính hỗ trợ thiết kế thuốc
Thiết kế thuốc sử dụng sự hỗ trợ máy tính (CADD) thường được áp dụng để dự đoán các chất ức chế tiềm năng có thể ngăn chặn hoạt động của enzyme/protein mục tiêu. Phương pháp này giúp làm giảm đáng kể thời gian và chi phí để phát triển một loại thuốc mới [5]. Việc xác định năng lượng (ái lực) tự do liên kết ∆G giữa enzyme và phối tử là một trong những yếu tố quan trọng nhất trong CADD bên cạnh việc ước tính dược động học và dược lý học các chất ức chế tiềm năng. Thông thường, để sàng lọc một số lượng lớn các chất, có thể lên đến nhiều triệu chất, CADD được thực hiện qua ba bước: 1) Sàng lọc ban đầu hàng nghìn/triệu hợp chất sử dụng phương pháp mối tương quan định lượng giữa cấu trúc và hoạt tính, docking phân tử; 2) Danh sách các hợp chất có ái lực cao sau đó sẽ được sàng lọc lại thông qua các phương pháp năng lượng liên kết tự do sử dụng nhiều tài nguyên tính toán hơn, như phương pháp năng lượng tương tác tuyến tính (LIE), hoặc kéo nhanh phối tử (FPL); 3) Các chất ức chế tiềm năng nhất cuối cùng sẽ được xác nhận thông qua việc xác định giá trị tuyệt đối của năng lượng tự do bằng phương pháp lượng tử năng lượng tự do nhiễu loạn, hay tích phân nhiệt động lực học. Ngoài ra, trí tuệ nhân tạo (AI) gần đây cũng bắt đầu được áp dụng nhiều để dự đoán chất ức chế tiềm năng.
Sàng lọc chất ức chế tiềm năng kháng SARS-CoV-2 Mpro sử dụng phương pháp docking phân tử
Docking phân tử thường được sử dụng để xác định nhanh vị trí và ái lực liên kết của các hợp chất thử với enzyme mục tiêu. Do sử dụng tài nguyên tính toán ít, docking phân tử có thể được sử dụng để nghiên cứu tương tác của hàng ngàn đến hàng triệu chất hướng tới mục tiêu. Ngay khi có cấu trúc thực nghiệm của SARS-CoV-2 Mpro, hàng loạt nghiên cứu docking phân tử đã được tiến hành nhằm dự toán chất ức chế tiềm năng cho protein mục tiêu này. Trong đó, có thể kể đến công bố của Thúy và cs (2020) [6] sử dụng phần mềm docking phân tử dự toán các hợp chất chiết xuất từ tỏi có khả năng ức chế SARS-CoV-2 Mpro. Các hợp chất được dự đoán có ái lực cao với Mpro đều là các hợp chất có chứa nguyên tố lưu huỳnh, như allyl disulfide, allyl trisulfide, allyl (E)-1-propenyl disulfide... Calligari và cs (2020) [7] cũng công bố kết quả nghiên cứu ái lực liên kết của các thuốc trong cơ sở dữ liệu nghiên cứu bệnh cúm (Influenze Research Database) sử dụng AutoDock Vina - một trong những phần mềm docking phân tử miễn phí phổ biến nhất.
Mô phỏng động lực học phân tử để đánh giá tiềm năng các chất ức chế SARS-CoV-2 Mpro
Do các mô phỏng docking phân tử thường sử dụng nhiều ràng buộc/xấp xỉ để đẩy nhanh tốc độ tính toán, kết quả cần được tinh chỉnh bằng cách sử dụng các phương pháp chính xác hơn [8]. Nghiên cứu của S.T. Ngo và cs (2020) [9] cho thấy, một số hợp chất tự nhiên trong thảo dược Việt Nam cũng có khả năng ức chế enzyme SARS-CoV-2 Mpro thông qua các mô phỏng tính toán nghiêm ngặt sử dụng phương pháp mô phỏng docking phân tử. Ngoài ra, các tính toán cũng cho thấy nếu có đột biến ở amino acid Glutamine ở vị trí 166, ái lực liên kết tự do của các phối tử tới SARS-CoV-2 Mpro sẽ thay đổi đáng kể (hình 2). Việc sàng lọc cơ sở dữ liệu các hợp chất bằng phương pháp tính toán cho thấy có 13 hợp chất liên kết mạnh với SARS-CoV-2 Mpro; đáng chú ý là delamanid, một loại thuốc chống lao được dự đoán là có thể ức chế SARS-CoV-2 Mpro rất mạnh. Vì cả COVID-19 và bệnh lao đều là bệnh phổi, nên delamanid có xác suất thích hợp để điều trị COVID-19 cao hơn các hợp chất dự đoán khác. Sử dụng kết hợp giữa docking phân tử và mô phỏng động lực phân tử (MD), Wang và cs (2020) [10] đã chỉ ra rằng, một số thuốc hiện hành có tiềm năng kháng SARS-CoV-2 Mpro như: arfilzomib, eravacycline, valrubicin, lopinavir và elbasvir. Đặc biệt, carfilzomib là một loại thuốc chống ung thư đã được phê duyệt hoạt động như một chất ức chế proteasome, có khả năng ức chế rất mạnh và ứng cử viên tốt thứ hai là eravacycline - kháng sinh nhóm tetracycline halogen hóa tổng hợp. Mới đây nhất, Gao và cs (2020) [11] đã sử dụng mô hình máy học để dự toán các hợp chất khác có thể sử dụng như là chất ức chế SARS-CoV-2 Mpro từ cơ sở dữ liệu của các thuốc hiện đang được lưu hành trên thị trường, trong đó có 314 chất ức chế SARS-CoV-2 thu được từ thực nghiệm đã được sử dụng nghiên cứu trong mô hình máy học, mở ra triển vọng tìm kiếm được các loại thuốc có khả năng ngăn chặn SARS-CoV-2 Mpro trong tương lai.
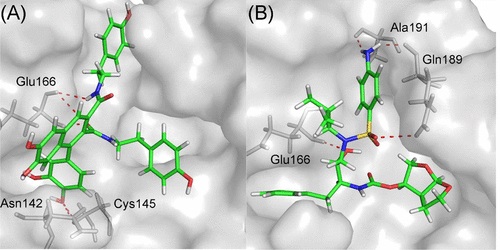
Hình 2. Tư thế liên kết của hệ SARS-CoV-2 Mpro + cannabisin A (A) và SARS-CoV-2 Mpro + darunavir (B), thu được thông qua mô phỏng động lực học phân tử [8].
Thay lời kết
Đại dịch COVID-19 xuất hiện đã thúc đẩy sự liên kết, hợp tác giữa các nhà khoa học trên thế giới nhằm tìm kiếm giải pháp chống lại đại dịch. Nhiều nhà hóa học tính toán và lý - sinh đã cố gắng tìm hiểu sâu hơn về hoạt động bên trong cũng như cơ chế phân tử của virus SARS-CoV-2, đồng thời tìm kiếm các phân tử nhỏ nhất có thể tương tác nhằm ức chế các protein của virus SARS-CoV-2. Nhờ sự phát triển mạnh mẽ của khoa học tính toán, đặc biệt là vật lý tính toán và hóa học tính toán, việc phát triển các loại thuốc tiềm năng hướng ức chế SARS-CoV-2 sẽ được tăng tốc, mở ra triển vọng điều chế các loại thuốc giúp phòng chống COVID-19 trong thời gian nhanh nhất.
TÀI LIỆU THAM KHẢO
[1] N. Doremalen, T. Bushmaker, D.H. Morris, M.G. Holbrook, A. Gamble, B.N. Williamson, A. Tamin, J.L. Harcourt, N.J. Thornburg, S.I. Gerber, J.O. Lloyd-Smith, E. de Wit, V.J. Munster (2020), "Aerosol and surface stability of SARS-CoV-2 as compared with SARS-CoV-1", N. Engl. J. Med., 382, pp.1564-1567.
[2] W. Dai, B. Zhang, H. Su, J. Li, Y. Zhao, X. Xie, Z. Jin, F. Liu, C. Li, Y. Li, F. Bai, H. Wang, X. Cheng, X. Cen, S. Hu, X. Yang, J. Wang, X. Liu, G. Xiao, H. Jiang, Z. Rao, L.K. Zhang, Y. Xu, H. Yang, H. Liu (2020), "Structure-based design of antiviral drug candidates targeting the SARS-CoV-2 main protease", Science, 368, pp.1331-1335.
[3] https://www.fda.gov/news-events/press-announcements/fda-approves-first-treatment-covid19?fbclid=IwAR3jElh3p4H0YrLtL0o92OE931R6ygixc2edh3zPX4E6SL4AbmMFNu19q8U.
[4] M.L. Holshue, C. DeBolt, S. Lindquist, K.H. Lofy, J. Wiesman, H. Bruce, S.I. Spitters, K. Ericson, S. Wilkerson, A. Tural, G. Diaz, A. Cohn, L. Fox, A. Patel, S.I. Gerber, L. Kim, S. Tong, X. Lu, S. Lindstrom, M.A. Pallansch, W.C. Weldon, H.M. Biggs, T.M. Uyeki, S.K. Pillai (2020), "First case of 2019 novel coronavirus in the United States", N. Engl. J. Med., 382, pp.929-936.
[5] G.R. Marshall (1987), "Computer-aided drug design", Ann. Rev. Pharmacol. Toxicol., 27, pp.193-213.
[6] B.T.P. Thuy, T.T.A. My, N.T.T Hai, L.T. Hieu, T.T. Hoa, H.T.P. Loan, N.T. Triet, T.T.V. Anh, P.T. Quy, P.V. Tat, N.V. Hue, D.T. Quang, N.T. Trung, V.T. Tung, L.K. Huynh, N.T.A. Nhung (2020), "Investigation into SARS-CoV-2 resistance of compounds in garlic essential oil", ACS Omega, 5, pp.8312-8320.
[7] P. Calligari, S. Bobone, G. Ricci, A. Bocedi (2020), "Molecular investigation of SARS-CoV-2 proteins and their interactions with antiviral drugs", Viruses, 12, p.445.
[8] S.T. Ngo, N.Q.A. Pham, L.T. Le, D.H. Pham, V.V. Vu (2020), "Computational determination of potential inhibitors of SARS-CoV-2 main protease", J. Chem. Inf. Model., 60, pp.5771-5780.
[9] S.T. Ngo, N.H. Minh, H.T.T. Le, Q.M. Pham, T.K. Vi, T.T. Nguyen, V. Van (2020), "Assessing potential inhibitors for SARS-CoV-2 main protease from available drugs using free energy perturbation simulations", RSC Adv., 10, pp.40284-40290.
[10] J. Wang (2020), "Fast identification of possible drug treatment of coronavirus disease-19 (COVID-19) through computational drug repurposing study", J. Chem. Inf. Model., 60, pp.3277-3286.
[11] K. Gao, D.D. Nguyen, J. Chen, R. Wang, G.W. Wei (2020), "Repositioning of 8565 existing drugs for COVID-19", J. Phys. Chem. Lett, 11, pp.5373-5382.